250
27
12
Areas of Interest
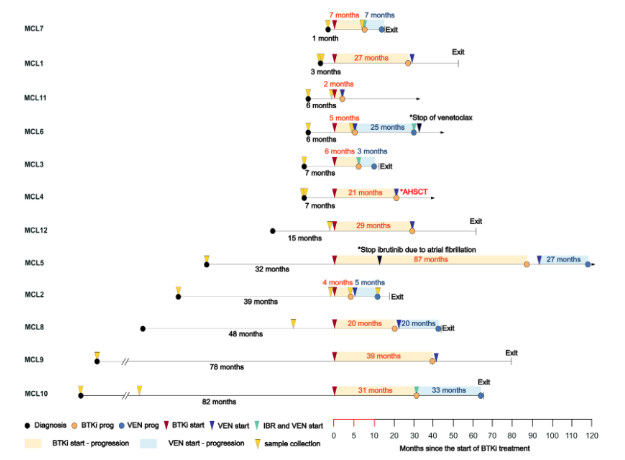
Resistance mechanisms and clonal dynamics in mantle celllymphoma treated with sequential BTKi and venetoclax therapy
This study investigated resistance mechanisms and clonal evolution in mantle cell lymphoma (MCL) patients treated sequentially with Bruton’s tyrosine kinase inhibitors (BTKi) and venetoclax. Using low-coverage whole genome sequencing (lcWGS), the researchers analyzed 21 tumor samples from 12 patients to identify genome-wide copy number alterations (CNAs) associated with treatment resistance. They discovered that samples collected after treatment had significantly more CNAs and that early progression on BTKi was associated with deletions or amplifications in key genes such as TRAF2, BIRC3, and ATM. Similarly, venetoclax resistance was linked to deletions in 9p21.3 affecting CDKN2A and other alterations involving NOTCH1, TRAF2, and CARD11. For mutation analysis of the tumor suppressor gene TP53, the study used Paragon Genomics’ CleanPlex® TP53 panel, which enabled targeted next-generation sequencing from formalin-fixed samples. This application of CleanPlex technology provided high-quality, reliable data on TP53 mutation status, supporting the broader genomic insights into resistance dynamics in MCL.
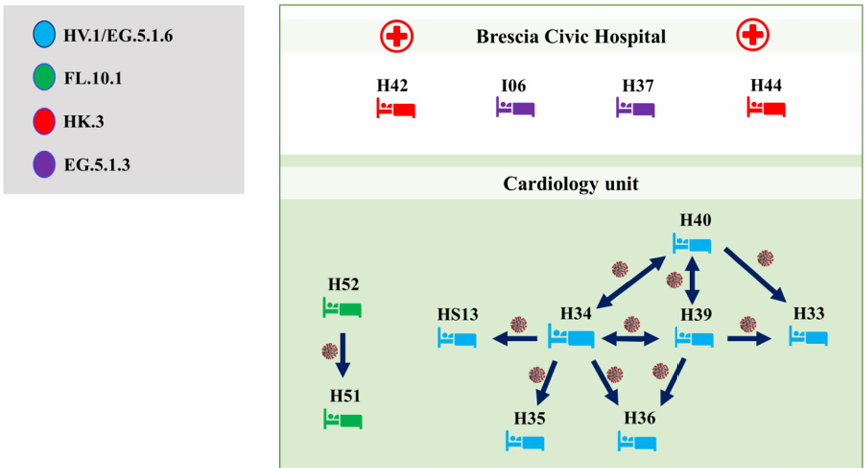
Tracking cryptic SARS-CoV-2 hospital outbreak through quasispecies analysis
The study aimed to use quasispecies analysis to track transmission of SARS-CoV-2, particularly in a hospital outbreak in Brescia Civic hospital from August to September 2023. Patients were molecularly tested for SARS-CoV-2 upon admission and those exhibiting respiratory symptoms were monitored through antigen screening. A total of 13 nasopharyngeal samples were collected. RNA extracted from those samples were sequenced using Paragon Genomics' CleanPlex multiplex PCR Research and Surveillance Panel for target enrichment, utilizing a streamlined multiplex PCR-based workflow to efficiently amplify selected genomic regions from extracted DNA. The indexed libraries were then sequenced using an Illumina platform, allowing for precise variant detection. The results demonstrated that nine patients hospitalized during the time of the study tested positive for SARS-CoV-2, all of whom tested negative in a molecular diagnostic test upon admission. Therefore, it was highly likely those patients acquired the virus during hospitalization. The subsequent epigenetic analyses combined with evaluation of the bottleneck dimension for 106 random couples allowed researchers to solve the outbreak and lay groundwork for systematic molecular surveillance in providing insights into transmission routes for newly emerging viral strains.
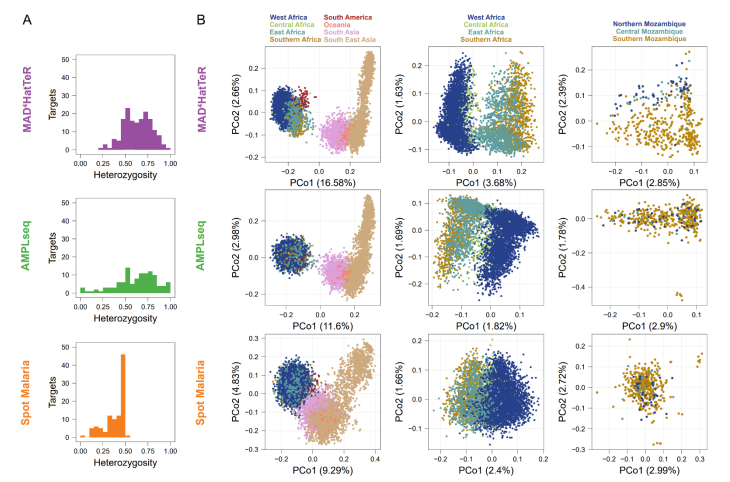
Sensitive and modular amplicon sequencing of Plasmodium falciparum diversity and resistance for research and public health
This research focused on developing MAD4HatTeR, a modular amplicon sequencing panel designed for Plasmodium falciparum genomic surveillance, enabling the detection of genetic diversity, drug resistance markers, and vaccine targets with high sensitivity and reproducibility. The study validated the panel across five laboratories, demonstrating its effectiveness in analyzing low-density infections and complex polyclonal samples. A crucial aspect of this work was the use of Paragon Genomics’ CleanPlex technology, which facilitated highly multiplexed PCR amplification of key genetic regions, along with offering a customizable approach for a wide scope project. This allowed researchers to efficiently sequence and analyze malaria-related genetic variations with improved sensitivity and specificity compared to traditional genotyping methods. By integrating targeted amplicon sequencing and a bioinformatics pipeline, the study provides a cost-effective and scalable tool for malaria research and public health surveillance, aiding in drug resistance monitoring and transmission tracking
Detection of Molecular Biomarkers in Fanconi Anemia Mucosa
The study investigates the usage of noninvasive screening for identification of molecular biomarkers in Fanconi anemia (FA), the most common cause of inherited bone marrow failure, using oral mucosa of patients to improve early detection of squamous cell carcinoma with the goal of developing molecular screening tools to fascilitate earlier detection of oral cancers. Using non-invasive brush biopsies from six oral sites, researchers employed the CleanPlex TP53 kit on the Illumina MiSeq to sequence TP53 exonic regions, detecting variants above 1% frequency. The study included 20 healthy controls and 17 FA patients. CleanPlex successfully identified 10 clinically significant TP53 variants in 33.3% of FA patients, while none were found in controls. This validation of non-invasive screening could enable better monitoring and earlier intervention in FA patients.
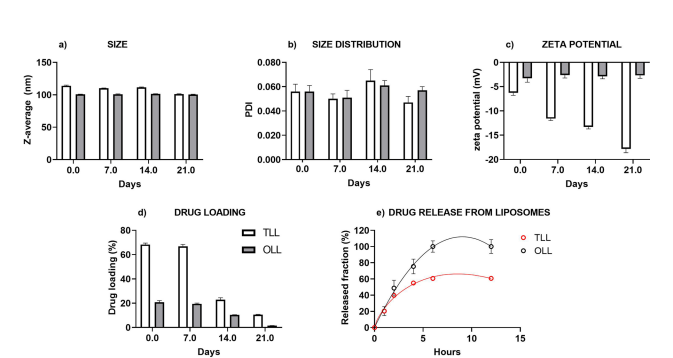
Synergistic antitumor effect of liposomal‑based formulations of olaparib and topotecan in primary epithelial ovarian cancer cells
The study investigated the effectiveness of combining liposomal formulations of olaparib and topotecan for treating primary epithelial ovarian cancer, particularly in BRCA1/2 wild-type cells. The methodology involved encapsulating olaparib (OLL) and topotecan (TLL) into liposomes to enhance drug delivery and targeting. The researchers evaluated the cytotoxic effects of these formulations on different ovarian cancer cell lines and healthy ovarian epithelial cells (HEOC) using various assays to measure cell viability and apoptosis. An important component of the study was the use of Paragon Genomics’ CleanPlex BRCA1 and BRCA2 kit, which enabled the researchers to accurately sequence the BRCA1 and BRCA2 genes. This technology provided comprehensive coverage of both exonic regions and 20bp into the adjoining intronic sequences, ensuring that no pathogenic mutations were overlooked. By confirming the BRCA wild-type status of the ovarian cancer samples, CleanPlex played a pivotal role in validating the effectiveness of the drug combinations in this specific genetic context. The results revealed that OLL and TLL formulations significantly improved the efficacy of these drugs, showing enhanced cytotoxicity in cancer cells while reducing toxicity to healthy cells. The combination of both drugs in liposomal form demonstrated a synergistic effect, leading to greater reductions in cancer cell viability compared to individual treatments. The study suggests that these liposomal drug formulations hold promise for more effective ovarian cancer treatment, particularly in cases resistant to traditional therapies.
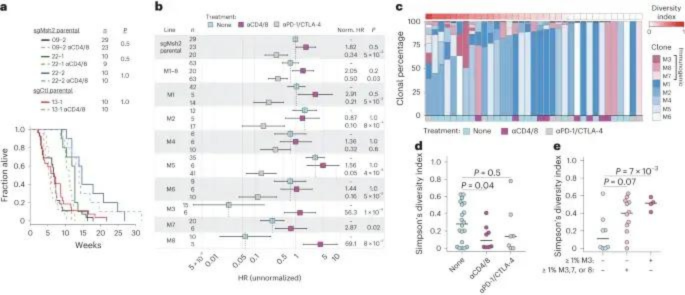
Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity
The Nature Genetics study investigates the role of mismatch repair deficiency (MMRd) in cancer immunogenicity, particularly in lung and colon cancers. Despite the high tumor mutational burden (TMB) typically associated with MMRd, the researchers found that these tumors did not exhibit increased immunogenicity or strong responses to immune checkpoint blockade (ICB) therapy. This was attributed to intratumor heterogeneity (ITH), where subclonal mutations fail to trigger effective immune responses. The findings suggest that clonal neoantigen burden, rather than overall TMB, is a better predictor of ICB response. Paragon Genomics CleanPlex UMI Custom Panel plays a key role in this research by enabling the accurate detection of clonal and subclonal mutations, providing insights into how mutations contribute to immune evasion and therapeutic outcomes.
Resistance mechanisms and clonal dynamics in mantle celllymphoma treated with sequential BTKi and venetoclax therapy
This study investigated resistance mechanisms and clonal evolution in mantle cell lymphoma (MCL) patients treated sequentially with Bruton’s tyrosine kinase inhibitors (BTKi) and venetoclax. Using low-coverage whole genome sequencing (lcWGS), the researchers analyzed 21 tumor samples from 12 patients to identify genome-wide copy number alterations (CNAs) associated with treatment resistance. They discovered that samples collected after treatment had significantly more CNAs and that early progression on BTKi was associated with deletions or amplifications in key genes such as TRAF2, BIRC3, and ATM. Similarly, venetoclax resistance was linked to deletions in 9p21.3 affecting CDKN2A and other alterations involving NOTCH1, TRAF2, and CARD11. For mutation analysis of the tumor suppressor gene TP53, the study used Paragon Genomics’ CleanPlex® TP53 panel, which enabled targeted next-generation sequencing from formalin-fixed samples. This application of CleanPlex technology provided high-quality, reliable data on TP53 mutation status, supporting the broader genomic insights into resistance dynamics in MCL.
Synergistic antitumor efect of liposomal‑based formulations of olaparib and topotecan in primary epithelial ovarian cancer cells
The study investigated the effectiveness of combining liposomal formulations of olaparib and topotecan for treating primary epithelial ovarian cancer, particularly in BRCA1/2 wild-type cells. The methodology involved encapsulating olaparib (OLL) and topotecan (TLL) into liposomes to enhance drug delivery and targeting. The researchers evaluated the cytotoxic effects of these formulations on different ovarian cancer cell lines and healthy ovarian epithelial cells (HEOC) using various assays to measure cell viability and apoptosis. Paragon Genomics CleanPlex BRCA1 and BRCA2 kit provided the coverage that researching tumor suppressor genes requires, targeting not only the exonic regions but also 20bp into the adjoining intronic sequences as well. That way, any pathogenic mutations would not escape notice. The results revealed that OLL and TLL formulations significantly improved the efficacy of these drugs, showing enhanced cytotoxicity in cancer cells while reducing toxicity to healthy cells. The combination of both drugs in liposomal form demonstrated a synergistic effect, leading to greater reductions in cancer cell viability compared to individual treatments. The study suggests that these liposomal drug formulations hold promise for more effective ovarian cancer treatment, particularly in cases resistant to traditional therapies.
Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity
This Nature Genetics study investigates the role of mismatch repair deficiency (MMRd) in cancer immunogenicity, particularly in lung and colon cancers. Despite the high tumor mutational burden (TMB) typically associated with MMRd, the researchers found that these tumors did not exhibit increased immunogenicity or strong responses to immune checkpoint blockade (ICB) therapy. This was attributed to intratumor heterogeneity (ITH), where subclonal mutations fail to trigger effective immune responses. The findings suggest that clonal neoantigen burden, rather than overall TMB, is a better predictor of ICB response. Paragon Genomics CleanPlex UMI Custom Panel plays a key role in this research by enabling the accurate detection of clonal and subclonal mutations, providing insights into how mutations contribute to immune evasion and therapeutic outcomes.
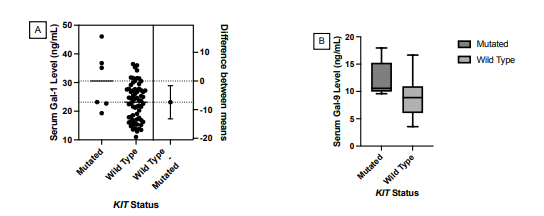
Correlational Analysis of Genetic Mutations and Galectin Levels in Breast Cancer Patients
This article explores the relationship between genetic mutations and galectin levels in individuals diagnosed with breast cancer. Galectins are proteins involved in various biological processes, including cancer progression, and their levels can be indicative of disease severity or prognosis. Genetic mutations, on the other hand, can influence cancer development and progression by altering cellular functions and pathways. In this study, researchers conducted a correlational analysis to investigate potential links between specific genetic mutations observed in breast cancer patients and the levels of galectins present in their tissues or bloodstream. By examining these correlations, the study aimed to identify potential biomarkers for breast cancer prognosis or treatment response. Paragon Genomics contributed to the study by providing our CleanPlex OncoZoom Cancer Hot-Spot Panel. Our targeted sequencing technologies were utilized to accurately identify genetic mutations in the breast cancer patients under investigation. This genetic data, when correlated with galectin levels, offered valuable insights into the molecular mechanisms underlying breast cancer and potentially identify novel biomarkers for diagnosis or treatment stratification.
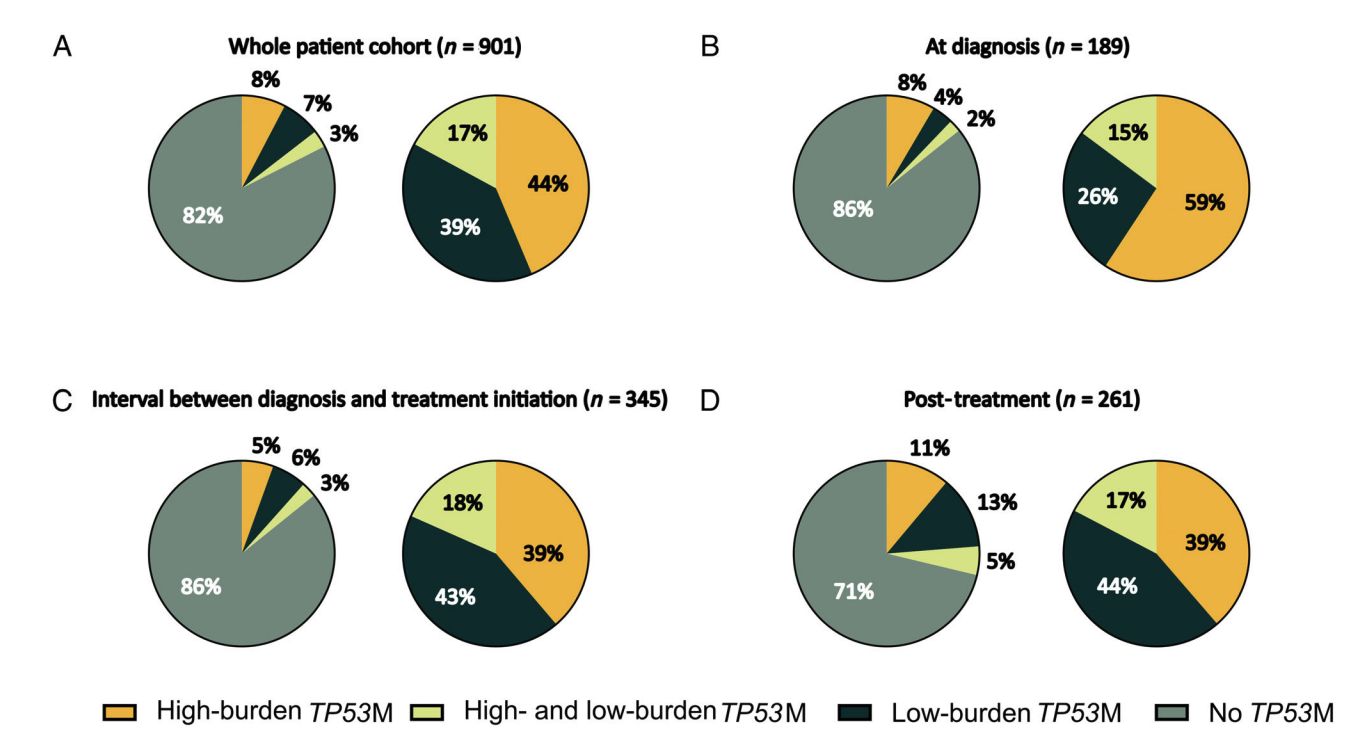
Low-burden TP53 mutations represent frequent genetic events in CLL with an increased risk for treatment initiation
Dr. Lazlo with fellow researchers performed a systematic study, delving into the significance of TP53 mutations in chronic lymphocytic leukemia (CLL) and their impact on chemo resistance. Leveraging Paragon Genomics' CleanPlex TP53 panel, they were able to identify 225 TP53 mutations in a real-world cohort of 991 patients with CLL, detecting somatic mutations with a frequency as low as 1%. The researchers were able to demonstrate that patients with sole low-burden TP53 variants represent more than 1/3 of patients with TP53 mutations and have an increased risk for treatment initiation, strengthening the need to redefine the threshold of TP53 variant reporting to below 10% in the routine diagnostic setting.

Detection of early seeding of Richter transformation in chronic lymphocytic leukemia
Prof. Campo published findings in Nature Medicine characterizing the genome of patients with Richter transformation (RT) of chronic lymphocytic leukemia (CLL). They discovered mutational signatures and driver alterations linked in the process of cancer evolution, and mechanisms that drive RT. 77 of those mutations were identified by Paragon’s Custom UMI NGS panel, with multiple of them classified as high confidence and detected at 0.5% LOD or lower (<0.1%), in which background was detected at 2-3 or more fold less. The panel was applied in a longitudinal patient study to characterize these mutational profiles in the onset of RT as correlated to specific drug therapies.
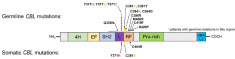
Molecular and phenotypic diversity of CBL mutated juvenile myelomonocytic leukemia
Dr. Stieglitz with fellow researchers at UCSF performed a systematic study of CBL mutations in juvenile myelomonocytic leukemia (JMML) with a custom 26-gene assay powered by Paragon Genomics’ CleanPlex technology and used it to correlate mutational burden with potential therapeutic regimens. Researchers were able to discover somatically acquired CBL mutations for the first time in JMML at the known hotspot loci, which has helped clarify why the disease is highly diverse in presentation and overall outcome for patients with CBL mutations

Single Circulating-Tumor-Cell-Targeted Sequencing to Identify Somatic Variants in Liquid Biopsies in Non-Small-Cell Lung Cancer Patients
Dr. Mouadh Barbirou and fellow researchers at the University of Missouri publish their findings that single circulating tumor cells (CTCs) detected by liquid biopsy can offer a robust tool for molecular profiling of cancer. Single CTCs were isolated and processed from non-small-cell lung cancer (NSCLC) patients via the RareCyte platform and 65 genes were targeted and sequenced using Paragon’s CleanPlex OncoZoom Cancer Hotspot NGS Panel. Somatic variants, particularly mutations in functional domains of seven critical oncogenes/tumor suppressor genes were identified and confirmed shared mutational signatures between the NSCLC patients. The study highlights the clear potential of CTC single-cell genomics in the field of precision oncology and its clinical value for diagnostics and therapy management.
Identification and Characterization of Effusion Tumor Cells (ETCs) From Remnant Pleural Effusion Specimens
Prof. Lowe from Stanford University along with colleagues from RareCyte, Inc performed a study to identify and characterize Effusion Tumor Cells (ETCs) from malignant pleural effusions in patients with metastatic lung adenocarcinoma. Pleural effusion specimens have not yet been routinely used for molecular testing because of their frequent low tumor fraction and sparse cellularity. In this study, researchers modified and applied a Circulating Tumor Cells testing method to examine malignancies from pleural effusion specimens. To obtain high-quality ETC libraries, they selected 5 representative patient samples each consisting of either a single cell or a small cell cluster. To generate Next-Generation Sequencing libraries, cell lysates were used as template with the CleanPlex OncoZoom Cancer Hotspot NGS Panel. Data Analysis and ETC Pool Variant Analysis was also performed. They retrieved high-quality sequencing results with great coverage uniformity. In this study, the authors developed a novel ETC-testing assay that detected epithelial malignancies in pleural effusions with 89.5% sensitivity and 100% specificity. Molecular profiling of ETCs showed promising concordance with their respective clinical molecular findings.
Functional CRISPR dissection of gene networks controlling human regulatory T cell identity
Prof. Alexander Marson from University of California, San Francisco (UCSF), CA, USA in collaboration with researchers from University of California, Berkeley, CA, USA, Technische Universität München (TUM), Munich, Germany and multiple renowned Cancer Immunotherapy institutes in San Francisco published findings in Nature Immunology showing CRISPR–Cas9 ribonucleoprotein (RNP) technology allows dissection of complex gene modules in primary human Treg cells through targeted gene perturbation studies. Using our CleanPlex custom panels, researchers were able to perform CRISPR editing efficiency check and show pooled Cas9 RNP screens link indels with phenotypic changes in human Treg cells. The overall study resulted in functional network maps which may help to guide future development of drug targets and design of Treg cell– based immunotherapies.
Somatic Mutations And T-Cell Clonality In Patients With Immunodeficiency
Dr. Satu Mustjoki and fellow researchers from University of Helsinki, Finland, in collaboration with the University Hospital and Computer Science Department of Aalto University School of Science, report an increase in somatic mutations in CD4+ and CD8+ T cells of patients with immunodeficiency. The team used a custom CleanPlex Panel to validate the low VAF somatic mutations to gain higher confidence in their findings.
Molecular assessment of pretransplant chemotherapy in the treatment of leukemia
Prof. Elliot Stieglitz‘s Lab at the UCSF Benioff Children’s Hospital developed a NGS assay powered by Paragon Genomics’ CleanPlex technology to measure the molecular burden of juvenile myelomonocytic leukemia (JMML). The researchers demonstrated that molecular testing can be helpful to distinguish between responders and nonresponders and should become an integral part of clinical care.
Tracking cryptic SARS-CoV-2 hospital outbreak through quasispecies analysis
The study aimed to use quasispecies analysis to track transmission of SARS-CoV-2, particularly in a hospital outbreak in Brescia Civic hospital from August to September 2023. Patients were molecularly tested for SARS-CoV-2 upon admission and those exhibiting respiratory symptoms were monitored through antigen screening. A total of 13 nasopharyngeal samples were collected. RNA extracted from those samples were sequenced using Paragon Genomics' CleanPlex multiplex PCR Research and Surveillance Panel for target enrichment, utilizing a streamlined multiplex PCR-based workflow to efficiently amplify selected genomic regions from extracted DNA. The indexed libraries were then sequenced using an Illumina platform, allowing for precise variant detection. The results demonstrated that nine patients hospitalized during the time of the study tested positive for SARS-CoV-2, all of whom tested negative in a molecular diagnostic test upon admission. Therefore, it was highly likely those patients acquired the virus during hospitalization. The subsequent epigenetic analyses combined with evaluation of the bottleneck dimension for 106 random couples allowed researchers to solve the outbreak and lay groundwork for systematic molecular surveillance in providing insights into transmission routes for newly emerging viral strains.
Sensitive and modular amplicon sequencing of Plasmodium falciparum diversity and resistance for research and public health
This research focused on developing MAD4HatTeR, a modular amplicon sequencing panel designed for Plasmodium falciparum genomic surveillance, enabling the detection of genetic diversity, drug resistance markers, and vaccine targets with high sensitivity and reproducibility. The study validated the panel across five laboratories, demonstrating its effectiveness in analyzing low-density infections and complex polyclonal samples. A crucial aspect of this work was the use of Paragon Genomics’ CleanPlex technology, which facilitated highly multiplexed PCR amplification of key genetic regions, along with offering a customizable approach for a wide scope project. This allowed researchers to efficiently sequence and analyze malaria-related genetic variations with improved sensitivity and specificity compared to traditional genotyping methods. By integrating targeted amplicon sequencing and a bioinformatics pipeline, the study provides a cost-effective and scalable tool for malaria research and public health surveillance, aiding in drug resistance monitoring and transmission tracking
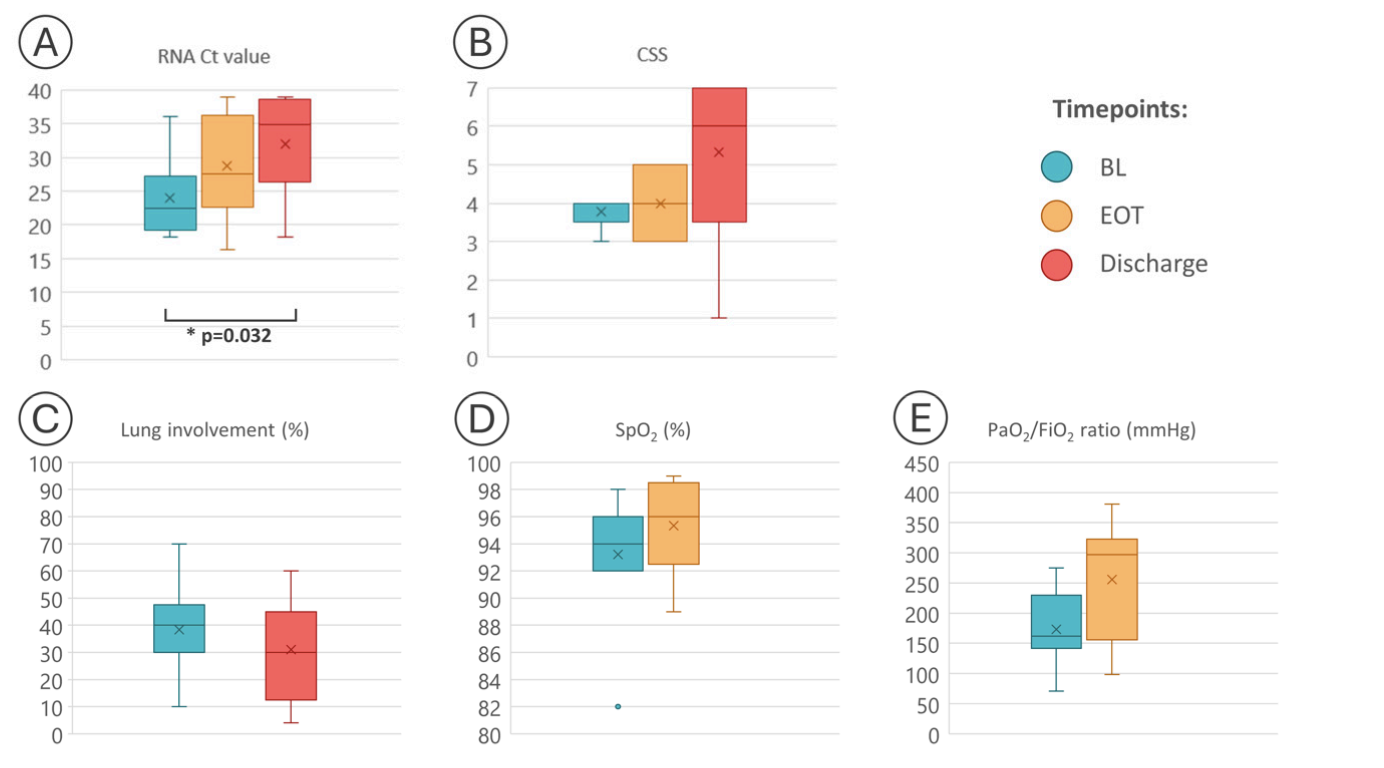
Clinico-Virological Outcomes and Mutational Profile of SARS-CoV-2 in Adults Treated with Ribavirin Aerosol for COVID-19 Pneumonia
This study retrospectively evaluated the effectiveness of ribavirin aerosol treatment in hospitalized adult patients with COVID-19 pneumonia. The research aimed to assess how ribavirin impacted the dynamics and clearance of the SARS-CoV-2 virus, as well as its effect on clinical outcomes and the virus mutational profile. Researchers utilized our CleanPlex® SARS-CoV-2 Panel to detect and measure RNA extracts of the virus, which allowed researchers to detect a significant decline in the virus cycle threshold (Ct) value, indicating reduced viral load in patients. Additionally, the study observed a mutational pattern in SARS-CoV-2 after ribavirin treatment in three out of five patients for whom whole-genome sequencing was available. This suggests that ribavirin may not only limit SARS-CoV-2 replication, leading to better clinical outcomes, but it may also contribute to the viral mutational spectrum.Thanks to our panel to accurately sequence and analyze viral RNA, researchers were able to find that ribavirin aerosol treatment may be effective in limiting SARS-CoV-2 replication and improving clinical outcomes in patients with COVID-19 pneumonia.
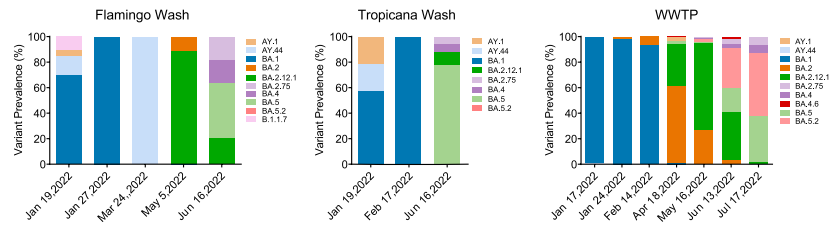
Environmental Surveillance of Flood Control InfrastructureImpacted by Unsheltered Individuals Leads to the Detection ofSARS-CoV‑2 and Novel Mutations in the Spike Gene
This article focuses on the environmental monitoring of flood control infrastructure, specifically examining areas affected by the presence of unsheltered individuals. Through this surveillance, researchers detected the presence of SARS-CoV-2, the virus causing COVID-19, as well as identified novel mutations in the spike gene. This study underscores the importance of environmental surveillance in understanding the spread of infectious diseases, particularly in vulnerable populations such as those experiencing homelessness. By monitoring areas where unsheltered individuals congregate, researchers can gain insights into the prevalence and transmission dynamics of pathogens like SARS-CoV-2. Paragon Genomics provided assistance by offering genomic sequencing solutions to analyze the genetic material collected during environmental surveillance. Using our CleanPlex SARS-CoV-2 Flex panel, our expertise in targeted sequencing helped researchers accurately identify and characterize SARS-CoV-2 and its genetic variations present in environmental samples. By leveraging our sequencing technologies, researchers gained insights into the prevalence, transmission dynamics, and genomic evolution of the virus in these specific environments, contributing to their understanding of COVID-19 spread and guiding public health responses.
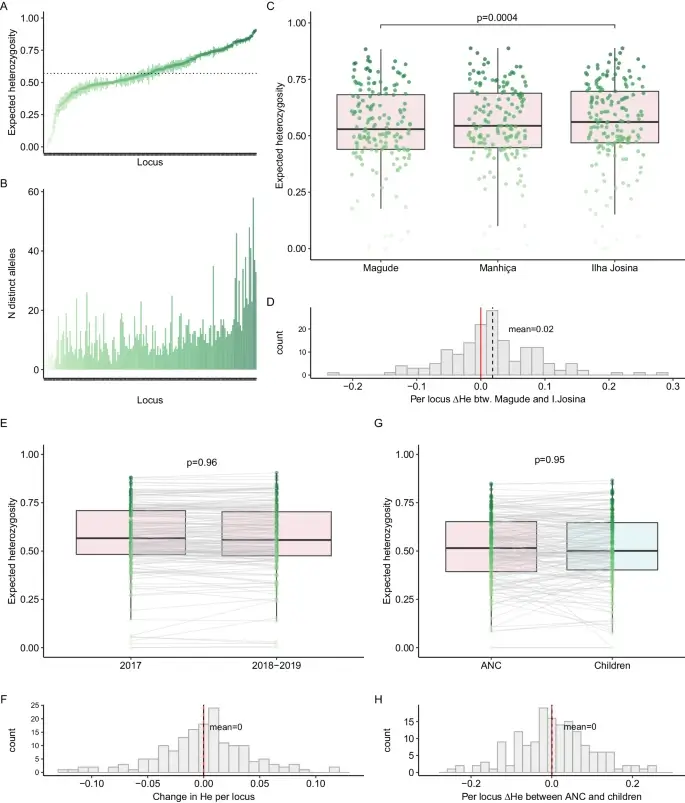
Genomic malaria surveillance of antenatal care users detects reduced transmission following elimination interventions in Mozambique
The research outlines how genomic surveillance, notably using the Paragon Genomics CleanPlex Malaria NGS Panel, at antenatal care visits in Mozambique demonstrates a decline in malaria transmission following elimination efforts. It highlights the consistent genetic diversity and drug resistance between malaria parasites in pregnant women and children, noting a significant genetic diversity reduction in targeted areas, indicative of reduced parasite populations. This approach underscores the potential of advanced genomic tools in enhancing malaria control and eradication strategies.
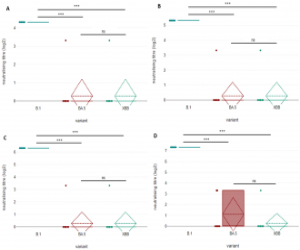
Omicron Sub-Lineage BA.5 and Recombinant XBB Evasion from Antibody Neutralisation in BNT162b2 Vaccine Recipients
Presently, the XBB recombinant variant, a hybrid of BA.2.10 and BA.2.75, which was first detected in August 2022, has given rise to apprehension due to its theoretically augmented infectivity and immune escape ability. In this study, authors tested the ability of this recombinant to evade vaccine-induced antibody neutralization compared to the ancestral lineage (B.1) and another co-circulating variant (B.1.1.529 BA.5) and were able to confirm that both BA.5 and XBB are significantly more immune-evasive, and to demonstrate that a specific vaccine (BNT162b2, BioNTech, Pfizer) only achieves a partial cross-neutralization of novel variants. The authors used Paragon’s CleanPlex SARS-CoV-2 FLEX NGS Kit for lineage identification of the clinical specimens, thereby showcasing the robustness of the FLEX degenerate primers for current SARS-CoV-2 variant identification capabilities.
Comparison of SNVs between Immunocompetent (IM+) and Immunocompromised (IM-)
COVID-19 Infection and Transmission Includes Complex Sequence Diversity
Dr Peter Zimmerman and colleagues at Case Western Reserve University were interested in characterizing the sequence diversity of the SARS-CoV-2 beyond the commonly reported majority consensus sequence in attempts to make new variant surges more easily detected and further understand and characterize infectious complexity. In this study, they used CleanPlex SARS-CoV-2 (panels 918010/918011) for whole genome sequencing of SARS-CoV-2 followed by variant calling on 254 clinical samples from COVID-19 patients to detect nucleotide changes outside of the consensus sequence region. The authors were able to detect a few mixed infection cases that occurred before the variant was publicly reported in the region, which may shed light on the usefulness for tracking SARS-CoV-2 beyond the consensus sequence for proper detection of new viral variants that may emerge during the pandemic.

External quality assessment of SARS-CoV-2-sequencing: An ESGMD-SSM (ESCMID Study Group for Genomic and Molecular Diagnostics - Swiss Society for Microbiology) pilot trial across 15 European laboratories
This first pilot study on external quality assessment of the SARS-CoV-2 whole genome sequencing was initiated by the ESGMD-SSM Study Group in Europe. Their aim was to build a framework between laboratories to improve COVID-19 surveillance sequencing. Ten samples with varying viral loads (Ct values = 19 to 28) were sent out to 15 different clinical laboratories who had free choice of library preparation kits, sequencing methods and bioinformatic analyses. The key aspects on which the individual centers were compared on were identification of 1) SNPs and indels, 2) Pango lineages, and 3) clusters between samples. The participating laboratories used a wide array of amplicon based library preparation methods including Illumina’s COVIDSeq test, ARTIC v3 workflow on both the Illumina and Oxford Nanopore sequencing technology and Paragon Genomics Inc.’s CleanPlex SARS-CoV-2 targeted amplicon technology. Most labs were able to generate whole genomes for all samples even with varying sequencing depth. There was a very good consensus regarding the majority of reporting criteria, but there were a few discrepancies in lineage and cluster assignment and variant calling. However, the CleanPlex SARS-CoV-2 amplicon sequencing technology (used in center 4), outperformed in almost all areas. CleanPlex’s results had the highest variant calling score in addition to the best accuracy, with no false positives (wrong) / false negative (missing) calls across all 10 samples (not even achieved by Illumina COVIDSeq, even with very deep sequencing).

A deletion in the N gene may cause diagnostic escape in SARS-CoV-2 samples
Prof. Sambri and his team at The Great Romagna Hub laboratory in Italy investigated a case study where routine diagnostic testing for COVID-19 showed discrepant results. After performing amplicon based Next Generation Sequencing with the CleanPlex SARS-CoV-2 kit to decode the viral genome, researchers were able to identify a consistent deletion in the region of the N gene as causal for diagnostic failure. As mutations in SARS-CoV-2 can confer ability to evade detection by RT-PCR, it is essential to continue surveying viral evolution and monitoring diagnostic accuracy via sequencing-based confirmation to mitigate false-negative PCR-based tests.
Paragon Genomics is proud to offer the CleanPlex SARS-CoV-2 FLEX Panel and Emerging Variants Panel Add-on specifically designed to contain additional components for robust and confident variant calling even when the virus mutates over time.
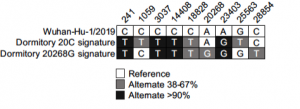
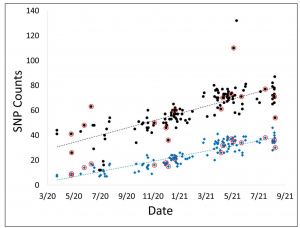
SARS-CoV-2 variant detection at a university dormitory using wastewater genomic tools
Prof. Oh from the University of Nevada along with his colleagues from the University of Arizona performed a WGS study to detect SARS-CoV-2 variants in wastewater collected at a university dormitory. Total RNA from wastewater was used to generate viral libraries using the CleanPlex SARS-CoV-2 FLEX Panel. Analysis of the sequencing results identified a mixture of two viral signatures that included clade 20C, with signature 20C mutations C1059T and G25563T, and another emerging viral strain corresponding to a 20A+20268G subclade. Compared to hybrid capture and other amplicon-based technologies, the lab was able to show libraries prepared using the CleanPlex SARS-CoV-2 FLEX Panel along with their genomic tools had the specificity and sensitivity to sequence single viral genomes. The investigators propose the implementation of cost-effective WGS, which enables public health authorities to definitively identify viral strains and manage local outbreaks compared to qPCR and digital PCR strategies which can be cost prohibitive.
Assessment of SARS-CoV-2 genome sequencing: quality criteria and low frequency variants
Prof. Claire Bertelli and her team at the Lausanne University Hospital performed a study to assess the sequencing quality criteria, especially for low frequency variants of the SARS-CoV-2 genome. The lab retrospectively investigated 647 SARS-CoV-2 genomes obtained over 10 tiled amplicons sequencing runs and identified eleven metrics associated with sequencing quality useful to easily implement quality enhancement measures. These amplicon libraries were constructed using the CleanPlex SARS-CoV-2 panel. Low frequency variants can result from PCR amplification errors, sample cross contaminations or presence of distinct SARS-CoV-2 genomes in the sample sequenced. To ensure the accuracy of SARS-CoV-2 genome sequences, it is essential to identify and distinguish PCR errors as well as potential sample cross-contaminations. Therefore, the team strongly recommends evaluating the proportion of low frequency variants, their prevalence in other SARS-CoV-2 samples from the same sequencing run, as well as their prevalence in all SARS-CoV-2 sequenced to date as a technical control before data submission to public health agencies and public databases. Their aim to provide simple quality control criteria for SARS-CoV-2 sequencing to prevent erroneous interpretation of low quality or contaminated data was accomplished.
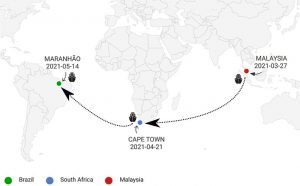
First reported cases of SARS-CoV-2 sub-lineage B.1.617.2 in Brazil: an outbreak in a ship and alert for spread
Prof. Luana Silva Soares and colleagues from the Department of Virology, Para state, Brazil reported the first instance of a SARS-CoV-2 B.1.617.2 (VoC) outbreak in an Asiatic origin trade ship which traveled from Malaysia and docked in Brazil. Nasopharyngeal swabs from 24 crew members were collected. The laboratory diagnosis was performed using RT-qPCR. Positive samples were used to perform Next Generation Sequencing using CleanPlex® SARS-CoV-2 Research and Surveillance Panel kits. SARS-CoV-2 genomes were obtained and classified as a B.1.617 lineage, confirmed by Pangolin lineage classification. Genetically, the B.1.617 lineage has a particular signature in the Spike protein, characterized by six marker mutations. In addition to those marker mutations, 21 other changes in the Spike protein were also observed in the NGS data. In conclusion, the lab reports the early detection of the SARS-CoV-2 lineage B.1.617 in Brazil. Since there are many factors still unclear about this VoC B.1.617.2, this single outbreak reinforces the necessity of continuous monitoring of travelers to follow the real-time spread of this new SARS-CoV-2 VoC, that could impact public health as well as immunization strategies.
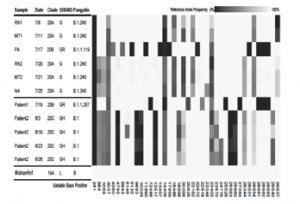
Genetic variant profiles of the sequenced SARS-CoV-2 relative to the Wuhan-Hu-1 reference genome.
Use of whole genome sequencing to investigate a cluster of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infections in emergency department personnel
Prof. Zimmerman from Case Western University and Prof. Donskey from the Cleveland VA Medical Centre performed a joint study to investigate a cluster of SARS-CoV-2 infections in 6 symptomatic emergency department personnel and 2 control COVID-19 patients. They used the CleanPlex SARS-CoV-2 Panel to perform viral whole genome sequencing to conduct this contact tracing investigation. Their results demonstrated that the suspected 6-person outbreak included 2 distinct transmission clusters and 1 unrelated infection. Their results suggest that SARS-CoV-2 variants initially acquired in the community were subsequently spread among hospital personnel. Hence, they concluded that such SARS-CoV-2 sequencing studies are needed to provide more definitive evidence regarding personnel as a source of transmission.
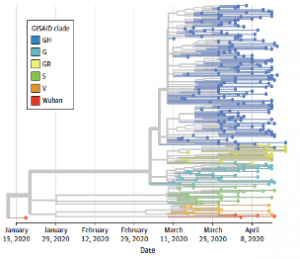
Genomic Epidemiology of SARS-CoV-2 Infection During the Initial Pandemic Wave and Association With Disease Severity
Prof. Rubin and Prof. Li from Case Western University along with their colleagues from Cleveland Clinic Ohio performed a cross-sectional study on 302 patients to examine the association of identified SARS-CoV-2 variants, virus clades, and clade groups with disease severity and patient outcomes. Whole genome sequencing of the virus was performed using the CleanPlex SARS-CoV-2 Panel followed by variant calling. Understanding of SARS-CoV-2 variants that alter disease outcomes are important for clinical risk stratification and provide important clues to the complex virus-host relationship. This study demonstrates a dynamic shift in SARS-CoV-2 clade diversity occurring very early in the pandemic. These findings suggest that SARS-CoV-2 clade assignment is an important factor that may aid in estimating patient outcomes.
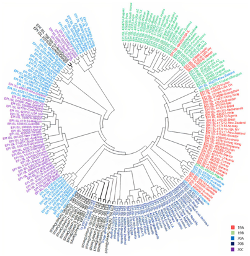
Whole Genome Sequencing and Phylogenetic Analysis of SARS‑CoV‑2 strains in Turkey
Prof. Hakan Akca from Pamukkale University, Turkey, performed whole genome sequencing on 15 COVID-19 positive patients using CleanPlex SARS-CoV-2 Panel . They identified 32 missense, 21 synonymous, and 4 non-coding allele mutations. Out of the missense mutations identified, 11 were rare missense mutations in the virus compared to the reference genome. Phylogenetic analysis revealed that most of the isolates were similar to European sequences. The phylogenetic tree constructed with all the complete SARS-CoV-2 genomes of Turkey showed that the viruses were spread nearly homogenous on eastern and western sides of Turkey.
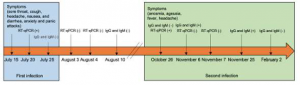
Evidence of SARS-CoV-2 reinfection within the same clade in Ecuador: A case study
Normally, reported SARS-CoV-2 reinfection cases are mainly seen in strains belonging to different clades however Prof. Zurita from Ecuador identified a case of reinfection within the same clade. Both, the infection and reinfection strains, were assigned as Nextstrain 20B, Pangolin lineage B.1.1 and GISAID clade O. These results were obtained by whole genome sequencing of the SARS-CoV-2 virus using the CleanPlex SARS-CoV-2 Panel. These results showed evidence that SARS-CoV-2 reinfection can occur within the same clade and previous infection to the virus cannot render complete immunity in all cases.
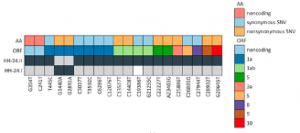
SARS-CoV-2 Reinfection in a Healthcare Worker Despite the Presence of Detectable Neutralizing Antibodies
Prof. Lütgehetmann from University Medical Center Hamburg-Eppendorf along with his colleagues from German Center for Infection Research performed a study to investigate the SARS–CoV–2 reinfection despite presence of detectable neutralizing antibodies in a female healthcare worker. Next generation sequencing (NGS) of the viral genome from the pharyngeal swab was performed using the CleanPlex SARS-CoV-2 Panel from the first and second episode of infection and were named #HH-24.I and HH-24.II, respectively. The NGS data revealed that the viral sequences from the initial virus variant in March (HH-24.I) and the variant in December (HH 24.II) belonged to pangoline lineages B.3 and B.1.177, respectively. In total, both sequences differed in 21 positions including two typical variations in spike proteins A222V and D614G. The team concluded that a better understanding about reinfection events may help to identify serological correlates of immunity.
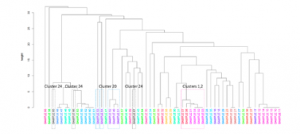
Utility of viral whole genome sequencing for institutional infection surveillance during the SARS-CoV-2 pandemic
Prof. Dien Bard and Prof. Gai from Children’s Hospital Los Angeles (CHLA) along with their colleagues from the Keck School of Medicine, USC performed whole genome sequencing (WGS) of 70 SARS-CoV-2 positive individuals using the CleanPlex SARS-CoV-2 NGS panel to evaluate institutional infection surveillance within a 9-month period. The researchers identified 25 potential clusters that warranted further exploration by WGS. Of the 25 suspected clusters analyzed, conclusive results were available in 23 cases from which they confirmed some relatedness in 14 clusters suspected by the contact tracing team including an outbreak within a unit. Importantly, WGS allowed them to rule out 9 highly suspicious clusters within the same units, demonstrating that these were not healthcare-associated infections. WGS analysis of SARS-CoV-2 isolates allowed the research team to monitor disease spread, outbreak dynamics, and infection hot spots within their institution and provided the capability to rule-out suspected clusters and confirm unrelated sources of infection. The team concluded that WGS analysis during the COVID-19 pandemic can be highly effective in a clinical setting as a complement to contact tracing efforts and will become increasingly important in identifying outbreaks.
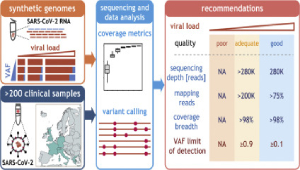
Recommendations for accurate genotyping of SARS-CoV-2 using amplicon-based sequencing of clinical samples
SOPHiA Genetics and researchers from 6 different institutes sequenced 200 clinical samples with Paragon Genomics’ CleanPlex SARS-CoV-2 Panel kit to generate what the authors claim as “much-needed recommendations” and guidelines for sequencing and genotyping of SARS-CoV-2 samples. They set criteria for accurate variant detection down to 10%, which enables sample tracing based on geographical origin and time of sampling. Additionally, the researchers identified dissemination patterns of the viral variants 20A.EU1 and 20A.EU2, which were linked to summer travel in 2020 across Europe. The results indicate high specificity with “virtual absence of sequencing reads mapping to the human transcriptome” and high uniformity that are trademarks of Paragon Genomics’ CleanPlex technology. Although more investigation is needed in the global fight against COVID-19, Paragon Genomics is proud to be helping the research community in its quest for better understanding of the SARS-CoV-2 genome.
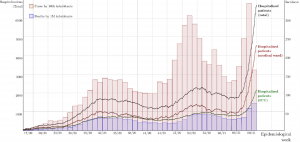
Detection of SARS-CoV-2 lineage P.1 in patients from a region with exponentially increasing hospitalization rates in February 2021, Rio Grande do Sul, Southern Brazil
Prof. Luis Barth and his colleagues from Federal University of Rio Grande do Sul and Hospital de Clínicas de Porto Alegre performed a study to look at the association of the emergence of the SARS-CoV-2 P.1 lineage and the rapid growth in hospitalization in the northern region of Brazil. Whole-genome sequencing of 68 patient specimens was performed using the CleanPlex SARS-CoV-2 panel and the data revealed that the previously undetected P.1 lineage was now detected and accounted for 88.9% of specimens collected from patients at a referral COVID-19 hospital. These findings raise concerns regarding a possible association between the lineage P.1 and rapid growth in cases and hospitalizations in the Rio Grande do Sul area.
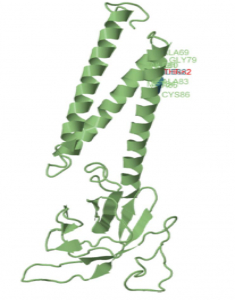
Emerging variants of concern in SARS-CoV-2 membrane protein: a highly conserved target with potential pathological and therapeutic implications
Prof. Biegel and Prof. Gai from Children’s Hospital Los Angeles (CHLA) along with their colleagues from the Keck School of Medicine, USC evaluated whole genome sequencing data from a cohort of 143,609 USA SARS-CoV-2 viral genomes to study the recent spike in frequency of the membrane protein (M) gene mutations in the SARS-CoV-2 genome. This cohort included 2,900 patients from CHLA whose whole genome data was generated using the CleanPlex SARS-CoV-2 NGS panel. The group reported a new Variant Of Concern with a signature mutation in the M gene, an otherwise overlooked but significant site of increasing numbers of mutations. In silico analysis revealed that the M protein structure is similar to that of the glucose transporter based upon which the M protein is thought to be involved in rapid viral proliferation, replication, and immune evasion. In the last few months, the incidence of some missense M gene mutations have increased over 100 fold and continue to increase. Hence, the researchers suggest that M gene mutations should be included in ongoing genomic surveillance efforts and warrants further evaluation.
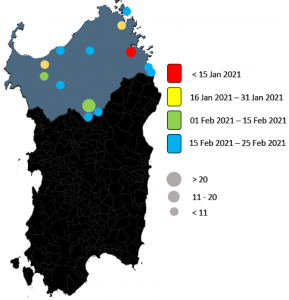
A straightforward molecular strategy to retrospectively investigate the spread of SARS-CoV-2 VOC202012/01 B.1.1.7 variant
Prof. Uzzau from University of Sassari along with his colleagues from Sassari University Hospital in Italy performed a retrospective study to investigate the spread of the SARS–CoV–2 Variant of Concern (VOC) 202012/01 B.1.1.7 in North Italy. They performed whole genome sequencing using the CleanPlex SARS-CoV-2 NGS Panel to confirm inconclusive diagnostic RT-PCR results. According to the Pangolin application, the analyses of this NGS data enabled the researchers to assign these viral genomes to SARS-CoV-2 lineages, confirming the presence of the B.1.1.7 variant. The CleanPlex SARS-CoV-2 NGS panel was able to ID the characteristic mutations along the whole SARS-CoV-2 viral genome including the mutations in the S gene encoding for Spike protein. Such variants represent a serious threat worldwide, thus conclusive and rapid methods are required for their identification.
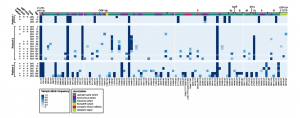
Persistent SARS-CoV-2 infection and increasing viral variants in children and young adults with impaired humoral immunity
Prof. Dien Bard and Prof. Gai from Children’s Hospital Los Angeles (CHLA) along with their colleagues from Stanford and Johns Hopkins performed a study in three patients with impaired humoral immunity who were persistently positive for SARS-CoV-2 infection. Whole-genome sequencing was performed using the CleanPlex SARS-CoV-2 Research and Surveillance NGS Panel and serological studies were performed to measure viral evolution and evidence of immune escape. Their work demonstrates that immunocompromised pediatric and young adult patients are susceptible to prolonged viral infections with continued virus shedding and mutation accumulation some with the potential to escape antibody neutralization. Their results highlight the need to reassess infection control precautions in the management and care of immunocompromised patients. Routine surveillance of mutations and evaluation of their potential impact on viral transmission and immune escape should be considered.
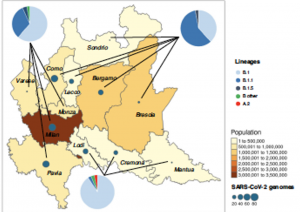
Genomic epidemiology of SARS-CoV-2 reveals multiple lineages and early spread of SARS-CoV-2 infections in Lombardy, Italy
Prof. Fausto Baldanti from Univeristy of Pavia, Italy and Prof. Carlo Federico Perno from Children’s Hospital, IRCCS, Rome, Italy along with their colleagues from University of Milan, Italy and ASST Grande Ospedale Metropolitano Niguarda, Milan, Italy performed a genomic epidemiology study of the SARS-CoV-2 genomes in Lombardy, Italy. In this study, they used an integrated approach comprising of both epidemiological and viral genetic data to reconstruct the pattern of the SARS-CoV-2 spread in this region. Whole-genome sequencing was performed using the CleanPlex SARS-CoV-2 Research and Surveillance NGS Panel for 346 SARS-CoV-2 strains obtained from individuals from various geographical areas in a time span of 2 months. In conclusion, this study contributed to the identification and circulation pattern of seven SARS-CoV-2 lineages in an area highly affected by COVID-19. In addition, they demonstrated the presence of local transmission clusters within three of those lineages, revealing that sustained community transmission was underway before the first COVID-19 case had been detected in Lombardy.
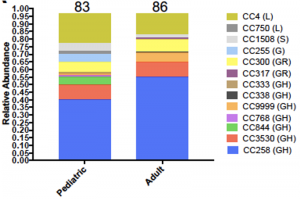
Early pandemic molecular diversity of SARS-CoV-2 in children
Prof. Paul Planet from Children’s Hospital of Philadelphia (CHOP) and Prof. Dien Bard and Prof. Gai from Children’s Hospital Los Angeles (CHLA) along with their colleagues from USC and UPenn performed a study in a pediatric population to track the molecular epidemiology of the SARS-CoV-2 virus locally in Philadelphia. They used the CleanPlex SARS-CoV-2 Research and Surveillance NGS Panel to perform whole genome sequencing followed by novel genotyping tools to quantifying the viral diversity on 169 SARS-CoV-2 genomes (83 genomes <21 years old) collected over three months. They show that the early pandemic and peak in Philadelphia were characterized by multiple, diverse, circulating viral variants, especially amongst children. Their analysis uncovered 13 major lineages that changed in relative abundance as cases peaked in mid-April. They conclude that no clear differences in clinical presentation were associated with the different virus lineages.

Detection of the new SARS-CoV-2 variant B.1.526 with the Spike E484K mutation in South America
Prof. Derly Andrade Molina from Espiritu Santo University and Prof. Ricardo Soto Rifo from University of Chile along with their colleagues performed an epidemiological surveillance and reported two sequences in South America of the new SARS-CoV-2 variant, B.1.526, harboring the immune escape-associated mutation E484K. These viral sequences of the new variant were obtained from two individuals with a negative RT-PCR test but were analyzed further by whole genome sequencing using the CleanPlex ® SARS-CoV-2 FLEX Panel. From all the mutations identified in this new variant, mutation E484K is located in the receptor binding domain of the spike protein hence being considered a variant of concern. Such variants can be transmitted faster and could be implicated in increased mortality and decreased vaccine efficiency. Therefore, the researchers conclude that genomic and immune surveillance are critical to determining the impact of this new variant in SARS-CoV-2 transmission and immunity dynamics.
High Prevalence of SARS-CoV-2 Genetic Variation and D614G Mutation in Pediatric Patients with COVID-19
Prof. Jennifer Dien Bard and Prof. Xiaowu Gai along with their colleagues from Children’s Hospital Los Angeles and Keck School of Medicine at USC, performed a study in pediatric patients infected with COVID-19 to study the genetic diversity of the SARS-CoV-2 viral genome. Whole genome sequencing was performed on extracted viral RNA from respiratory specimens collected on all SARS-CoV-2 positive children (n=141) using Paragon Genomics CleanPlex SARS-CoV-2 Research and Surveillance NGS Panel. They analyzed the relationships between viral genetic variants and clinical characteristics in children. Genomic evaluation demonstrated greater than expected genetic diversity, presence of the D614G mutation in the viral spike protein, increased mutation rate, and evidence of multiple introductions of SARSCoV-2 into Southern California. Their findings suggest a possible association of phylogenetic clade 20C with severe disease, but small sample size precludes a definitive conclusion. The researchers suggest a further multi-institutional genomic evaluation is needed and can have implications on infection control practices.

PCORI Health Care Horizon Scanning System: Horizon Scanning COVID-19 Supplemental High Impact Report
The PCORI Health Care Horizon Scanning System has recently made a notable mention of Paragon Genomics to the likes of companies such as Illumina and Helix to be instrumental in developing several NGS assays for COVID-19 testing. The Horizon Scanning System identifies technology and systems innovations that could disrupt or cause significant shifts in health care. The Horizon scanning system can identify new (and new uses of existing) lab tests and procedures, public health and health promotion activities, and therapeutic interventions. They have issued a high impact report this September 2020 reporting several companies like Illumina, Paragon Genomics, Helix and others that have developed several NGS assays that can employ deep sequencing and computational methods to detect nucleic acids of the coronavirus. Such tests can amplify and detect sequences from the whole viral genome or several genomic regions. In their words ”NGS assays purportedly offer high throughput, enabling 2000 to 3000 samples to be processed in about 12 hours, with accuracy comparable to that of standard PCR tests and these assays might also be used in epidemiology studies to track the spread of coronavirus or to screen individuals who are planning to return to work or school.”
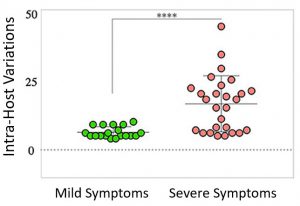
Within-host diversity of SARS-CoV-2 in COVID-19 patients with variable disease severities
Prof. Hadi M. Yassine from Qatar University in Doha, Qatar in collaboration with other researchers investigated the within-host SARS-CoV-2 virus diversity of 19 and 27 COVID-19 patients with mild and severe clinical manifestations respectively. Using our CleanPlex® SARS-CoV-2 Panel kit they performed targeted virus genome sequencing and reported patients with severe symptoms exhibited a significantly higher number of variants in coding and non-coding regions compared to mild cases. Their analysis also revealed higher prevalence of certain variants among severe cases only. Most importantly, severe cases exhibited significantly higher within-host diversity compared to mild cases. These observations helped them conclude that within-host diversity of the virus might play a role in the development of severe disease outcomes in COVID-19 patients.

Characterization of local SARS-CoV-2 isolates and pathogenicity in IFNAR−/- mice
Prof. Aykut Özkul from Ankara University in Turkey performed a study to isolate SARS-CoV-2 viruses circulating in Anatolia, and to investigate virus propagation in frequently-used cells and experimental animals. Using our CleanPlex® SARS-CoV-2 panel for their amplicon-based next generation sequencing, they identified two local SARS-CoV-2 virus isolates, and investigated in vitro growth dynamics in frequently-used Vero E6 cells and identified IFNAR−/− mice as a potential animal model for SARS-CoV-2 experiments.
Pediatric COVID-19 in Southern California: clinical features and viral genetic diversity
Prof. Xiaowu Gai and Prof. Jennifer Dien Bard from Children’s Hospital Los Angeles (CHLA) recently published a paper exploring clinical features and SARS-CoV-2 genetic variation in children presenting with COVID-19. They observed diverse clinical presentations and identified association between disease severity, viral load and age. By using CleanPlex SARS-CoV-2 Research and Surveillance NGS Panel, they were able to conduct highly sensitive and full-genome interrogation of the SARS-CoV-2 virus using amplicon sequencing on Illumina platforms.
Sensitive, highly multiplexed sequencing of microhaplotypes from the Plasmodium falciparum heterozygome
Prof. Bryan Greenhouse, Dr. Sofonias Tessema and fellow researchers from Chan Zuckerberg Biohub, University of San Francisco, USA in collaboration with researchers from University of Massachusetts, Brown University, Clinton Health Access Initiative, and Centro de Investigação em Saúde de Manhiça (CISM) of Mozambique demonstrated that targeted NGS sequencing with a custom CleanPlex Panel is a flexible, low cost, “promising tool for studying the transmission dynamics of P. falciparum” infections (Malaria), without the need for WGA, describing that in fact, their CleanPlex approach “outperformed whole genome sequences obtained from 50 times more total reads”. They further go on to note that their custom CleanPlex Panel “demonstrated high specificity regardless of the proportions of strains in the mixture (true positive rate = 99.8)… [and] sensitivity of the method was high across a wide range of parasite densities.”
The origin of SARS-CoV-2 in Istanbul: Sequencing findings from the epicenter of the pandemic in Turkey
Prof. Dinler-Doganay’s Lab at Istanbul Technical University, Prof. Doganay and his colleagues at University of Health Sciences, Istanbul, Turkey and Dr. Ilker Karacan from Science and Advanced Technology Research Center, Istanbul, Turkey reported the SARS-CoV-2 viral genomes circulating in Istanbul for the first time. By using Paragon Genomics’ CleanPlex SARS-CoV-2 Research and Surveillance Panel, amplicon-based next-generation sequencing approach was successfully applied to generate near-complete genomes with an average depth of 2616. All three viral genomes carried the D614G variant (G clade according to GISAID classification) with implications for the origin of a spread.
Highly sensitive and full-genome interrogation of SARS-CoV-2 using multiplexed PCR enrichment followed by next-generation sequencing
Prof. Xiaowu Gai and Prof. Jennifer Dien Bard from Children’s Hospital Los Angeles (CHLA), Paragon Genomics R&D teams led by Dr. Zhitong Liu, Prof. Alexander Urban from Stanford University and scientists from MGI / BGI-ShenZhen collaborated on sequencing COVID-19 patient samples using Paragon Genomics’s CleanPlex SARS-CoV-2 Panel for highly sensitive and full-genome interrogation of the SARS-CoV-2 virus using multiplexed PCR enrichment followed by next-generation sequencing.
Detection of Molecular Biomarkers in Fanconi Anemia Mucosa
The study investigates the usage of noninvasive screening for identification of molecular biomarkers in Fanconi anemia (FA), the most common cause of inherited bone marrow failure, using oral mucosa of patients to improve early detection of squamous cell carcinoma with the goal of developing molecular screening tools to fascilitate earlier detection of oral cancers. Using non-invasive brush biopsies from six oral sites, researchers employed the CleanPlex TP53 kit on the Illumina MiSeq to sequence TP53 exonic regions, detecting variants above 1% frequency. The study included 20 healthy controls and 17 FA patients. CleanPlex successfully identified 10 clinically significant TP53 variants in 33.3% of FA patients, while none were found in controls. This validation of non-invasive screening could enable better monitoring and earlier intervention in FA patients.
NOTCH3 Variants in Patients with Suspected CADASIL
Dr. Gorukmez from the Department of Medical Genetics and Neurology in Turkey has published findings investigating genetic variants of NOTCH3 in individuals with suspected cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), which is a rare hereditary form of cerebral small vessel disease. The researchers utilized Paragon Genomics’s CleanPlex technology to generate a custom targeted sequencing panel for analyzing patient samples isolated from blood and accurately identifying pathogenic variants in the NOTCH3 gene. By leveraging the Sophia DDM platform, the team successfully detected 30 different disease-causing variants, including 17 novel variants, in the patient group. These findings contribute valuable insights into the diagnosis and management of CADASIL, highlighting the significance of understanding the genetic basis of CADASIL and its potential impact on personalized medicine approaches.
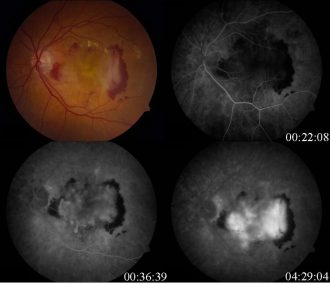
An assessment of prevalence of Type 1 CFI rare variants in European AMD, and why lack of broader genetic data hinders development of new treatments and healthcare access
Age related macular degeneration (AAMD) risk is associated with CFI (human complement factor I), but it remains unclear how variant prevalences might differ by ethnicity. To evaluate the prevalence of Type 1 CFI rare variant genotypes, targeted sequencing using Paragon’s CleanPlex technology was performed over the CFI coding region plus 50bp on either side of the intron/exon boundaries to determine VAF. Different ethnicities displayed markedly varying degrees of CFI variant prevalence, which further reinforces the need to generate more multi-ethnic AAMD datasets to better understand the impact of CFI genetic diversity on disease risk, and contribute to the emergence of novel tailored treatments.
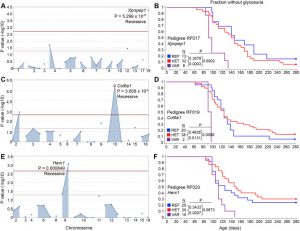
Modulation of autoimmune diabetes by N-ethyl-N-nitrosourea- induced mutations in non-obese diabetic mice
Modulation of autoimmune diabetes by ENU-induced mutations in Non-Obese Diabetic mice
Dr. Bruce Beutler and colleagues from UT Southwestern Medical Center in collaboration with Dr. Lucienne Chatenoud from Université de Paris were interested in clarifying specific genes that could be linked to type 1 diabetes. Using a plethora of mouse models, they applied custom panels from Paragon Genomics for loci detection and Ion Torrent sequencing on over 800 mice from 14 pedigrees bearing ~600 coding/splicing changes and identified several mutations that either accelerated, delayed, or suppressed type 1 diabetes development in their models, which may provide new insights to understanding human susceptibility to type 1 diabetes.
The role of genetic mutations in intrahepatic cholestasis of pregnancy
Dr. Gültekin Adanaş Aydın, Dr. Gülten Özgen, and Dr. Orhan Görükmez from Bursa Yuksek Ihtisas Training and Research Hospital, Bursa, Turkey published a paper about the role of genetic mutations in intrahepatic cholestasis of pregnancy using a CleanPlex custom panel.
Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome
Prof. José A.Sánchez-Alcázar and fellow researchers from University of Pablo de Olavide, Spain collaborated with researchers from Department of Neurology, Uniklinikum C. G. Carus, Dresden, Germany and Instituto de Salud Carlos III, Spain and used CleanPlex Mitochondrial Disease Panel to study parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome.
Building phylogenomic data sets using highly multiplexed amplicon sequencing
Dr. Scott Geib and fellow researchers at the USDA-Agricultural Research Service developed new approach for building phylogenomic data sets using highly multiplexed amplicon sequencing powered by Paragon Genomics’ CleanPlex technology. The work provided unparalleled resolution of phylogenetic relationships in fruit flies.
About Paragon Genomics
Our mission is to provide high-quality target enrichment solutions to the genomics community to advance precision medicine and research.
At Paragon Genomics, we are a passionate group dedicated to providing cutting-edge technologies and solutions for the global genomic research and application community. Our goal is to move precision medicine forward. The team is composed of industry veterans and academic experts who previously worked with well-known industry-leading companies and institutions such as ThermoFisher Scientific, Life Technologies, PerkinElmer, Beckman Coulter, Affymetrix, Novartis, Applied Biosystems, Siemens Healthcare, Stanford and Harvard.
Paragon Genomics’ leadership team proudly graduated from the California Life Sciences Institute’s (CLSI) Spring 2016 FAST Advisory Program. We were also honored to be accepted into the renowned StartX Fall 2016 Accelerator Program.