Product Description
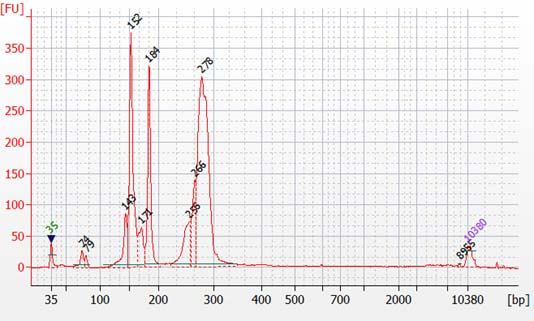
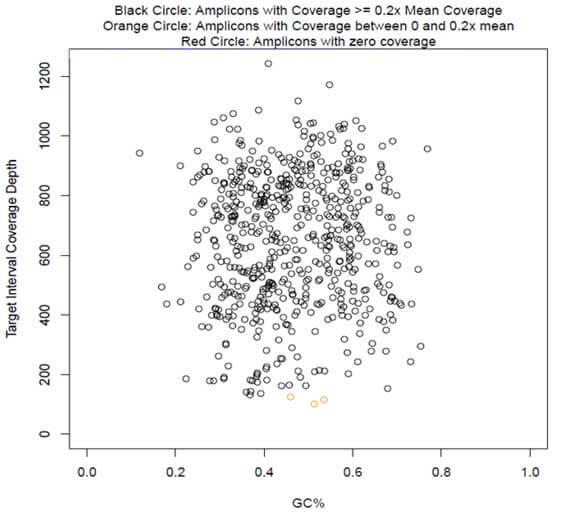
The CleanPlex OncoZoom Cancer Hotspot Kit is a multiplex PCR-based targeted resequencing assay designed for rapid cancer profiling of somatic mutations across the hotspot regions of 65 oncogenes and tumor suppressor genes. Starting with just 100 pg of high-quality genomic DNA, sequencing-ready libraries can be prepared using a single-tube workflow in just 3 hours. The panel is optimized to deliver data with high on-target performance and high coverage uniformity to ensure efficient use of sequencing reads.
Highlights
- Relevant Gene Content
Target 2,900+ hotspots in 65 genes with known cancer associations - Fast, Single-Tube Workflow
Generate sequencing-ready libraries in just 3 hours using a three-step, single-tube protocol - Superb Performance
Prepare high-quality NGS libraries with excellent on-target performance using CleanPlex Technology to enable efficient use of sequencing reads and reduce costs - Compatible with difficult samples including single cells
Short amplicon size design works well with fragmented DNA from FFPE, ctDNA from blood cfDNA, DNA from single circulating tumor cells and other clinical samples; The specific user guide for single cell can be found here.
The CleanPlex OncoZoom Cancer Hotspot Kit contains CleanPlex Multiplex PCR Primers and CleanPlex Targeted Library Kit. CleanPlex Indexed PCR Primers and CleanMag® Magnetic Beads are ordered separately to complete the workflow from input DNA to sequencing-ready NGS libraries.
Paragon Genomics has partnered with VarSome Clinical to provide a full solution for fast and accurate variant discovery, annotation, and interpretation of next-generation sequencing data for targeted gene panels. Click here to find out more!
Storage Temperature
Store at -20 °C.
For Research Use Only. Not for use in diagnostic procedures.